Bendrumas
Terminas pigmentinis retinitas (RP) nustato genetinių ligų grupę, kuriai būdinga progresuojanti tinklainės degeneracija.

Retinitis pigmentosa yra tinklainės distrofija, kuriai būdingas laipsniškas fotoreceptorių praradimas ir pigmento epitelio disfunkcija. Tai reiškia, kad tinklainė palaipsniui mažina jos gebėjimą per regos nervą perduoti vaizdinę informaciją į smegenis.
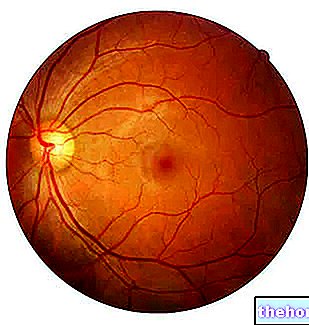
Patologinis procesas prasideda tinklainės pigmento epitelio pakitimais. Plečiantis retinitui, retėja kraujagyslės, tiekiančios tinklainę, kurios atrofuojasi. Ištyrus dugną, vizualiai aptinkamos būdingos nuosėdos. Tinklainės pigmentas ( taigi ir pavadinimas nuo ligos). Atrofiniai pokyčiai ir pažeidimai taip pat gali apimti regos nervą, o palaipsniui žūsta šviesai jautrios tinklainės ląstelės.
Pigmento retinito paveikti pacientai iš pradžių patiria regėjimo problemų, ypač prastai apšviestoje aplinkoje, ir skundžiasi periferinio regėjimo lauko susiaurėjimu. Centrinis regėjimas yra apsaugotas iki vėlesnių ligos stadijų, o galutinis rezultatas gali labai skirtis: daugelis žmonių, sergančių pigmentiniu retinitu, visą gyvenimą išsaugo ribotą regėjimą, o kiti visiškai praranda regėjimą.
Pigmentinis retinitas yra paveldima liga, kurią daugiausia sukelia genetiniai pokyčiai, perduodami iš vieno ar abiejų tėvų. Genetinio defekto tipas lemia, kurios tinklainės ląstelės labiausiai susijusios su sutrikimu, ir leidžia klinikiniu požiūriu atskirti skirtingas sąlygas. Iki šiol buvo nustatyta daugiau nei 50 skirtingų genetinių defektų, susijusių su retinitu pigmentosa. Anomalijos gali būti perduodamos iš tėvų palikuonims per vieną iš trijų paveldėjimo modelių: autosominis recesyvinis, autosominis dominuojantis arba heterosominis recesyvinis (susietas su X arba susietas su X).
Simptomai
Daugiau informacijos: Pigmentinės retinito simptomai
Retinitas pigmentosa dažniausiai pasireiškia paaugliams ir jauniems suaugusiems. Simptomai dažnai pasireiškia nuo 10 iki 30 metų, tačiau diagnozė gali būti nustatyta ankstyvoje vaikystėje arba daug vėliau.
Ankstyvieji pigmentinio retinito simptomai gali būti:
- Sunku matyti naktį (naktinis aklumas) arba esant silpnam apšvietimui
- Lėtas prisitaikymas nuo regėjimo tamsoje iki matymo šviesoje ir atvirkščiai;
- Regėjimo lauko susiaurėjimas ir periferinio regėjimo praradimas;
- Jautrumas šviesai ir blizgesiui.
Kai kurie simptomai priklauso nuo dalyvaujančių fotoreceptorių tipo. Strypai yra atsakingi už juodai baltą matymą, o kūgiai leidžia atskirti spalvas.
Daugeliu atvejų retinitis pigmentosa pirmiausia pažeidžia lazdeles. Tačiau sparčiai besivystančiose formose kūgiai taip pat gali būti paveikti ankstyvoje stadijoje.
Strypai sutelkti išorinėse tinklainės dalyse ir yra aktyvuojami dėl silpnos šviesos, todėl jų degeneracija veikia periferinį ir naktinį matymą. Jei yra kūgių, galite prarasti spalvų suvokimą ir centrinį regėjimą.
Dalyvaujančių fotoreceptorių dominavimą lemia ypatingas paciento genetinės sudėties defektas.
Dažnai pirmasis pigmentinio retinito simptomas yra naktinis aklumas (arba nocthalopia). Kai kurie žmonės mano, kad jiems reikia vis daugiau laiko prisitaikyti prie šviesos skirtumų, kai jie pereina iš gerai apšviestos vietos į tamsesnę. Tipiška regėjimo praradimo forma sukelia periferinio regėjimo susiaurėjimą (tunelio ar teleskopo regėjimas); šis modelis vadinamas žiedine skotoma. Kartais šio reiškinio gali trūkti ankstyvosiose stadijose, tačiau jis pastebimas, kai asmuo dažnai užkliūva už objektų arba patenka į automobilio avariją. Kai regos praradimas apima centrinę tinklainės sritį (dar vadinamą geltonosios dėmės distrofija) patiria sunkumų skaitydami ir atlikdami išsamius darbus, kuriems reikia sutelkti dėmesį į vieną objektą, pavyzdžiui, sriegiuoti siūlą per adatos akį Daugelis pacientų praneša, kad mato šviesos blyksnius (fotopsiją), dažnai apibūdinamus kaip mažos, mirgančios ir mirksinčios lemputės.
Ligos progresavimo greitis ir regos praradimo laipsnis kiekvienam asmeniui skiriasi. Kai kurie kraštutiniai atvejai gali greitai išsivystyti per du dešimtmečius, kiti - lėta eiga, kuri niekada nesukelia visiško aklumo. Ankstyvas pasireiškimas pasireiškia sunkesnėmis pigmento retinito formomis, o lengvesnės būklės (pvz., Autosominis dominuojantis) pacientams liga gali išsivystyti penktą ar šeštą gyvenimo dešimtmetį. Šeimose, kuriose yra su X susietas pigmentinis retinitas, vyrai serga dažniau moterys ir sunkiau; moterys, priešingai, perduoda genetinę charakteristiką (jos neša pakitusią geną X chromosomoje) ir rečiau pasireiškia sutrikimo simptomai.
Komplikacijos
Pigmento retinitas ir toliau progresuos, nors ir lėtai. Tačiau visiškas aklumas yra retas, tačiau gali labai sumažėti periferinis ir centrinis regėjimas.
Pacientams, sergantiems retinitu pigmentosa, ankstyvame amžiuje dažnai atsiranda tinklainės patinimas (geltonosios dėmės edema) arba katarakta. Šios komplikacijos gali būti gydomos, jei jos trukdo regėjimui.
Susijusios ligos
Paprastai pacientas, sergantis pigmentiniu retinitu, neturi kitų sutrikimų ir šiuo atveju mes kalbame apie „nesindrominį“ arba paprastą pigmentinį retinitą. Tačiau kai kuriems sindromams būdingi kai kurie klinikiniai šios akių ligos simptomai; labiausiai paplitęs yra Usherio sindromas, kuris pasireiškia maždaug 10–30% visų pacientų, sergančių pigmentiniu retinitu ir yra susijęs su įgimtu ar progresuojančiu klausos praradimu. Tačiau įgimtoje Leberio amaurozėje vaikai per pirmuosius šešis gyvenimo mėnesius gali apakti arba beveik apakti.Kitos ligos, susijusios su retinitu pigmentosa, yra Bardet-Biedl sindromas ir Refsum liga.
Priežastys
Ligą gali sukelti daugybė genetinių defektų: iš tikrųjų yra keletas genų, kurie, paveikti pakitimų, gali sukelti pigmento retinito fenotipą. Paprastai jie koduoja baltymus, dalyvaujančius transdukcijos kaskadoje, kuri leidžia matyti, veiksniai, lemiantys ląstelių transkripciją (kurie siunčia klaidingus pranešimus į tinklainės ląsteles) arba elementams, sudarantiems fotoreceptorių struktūrą. Paveldimos genų mutacijos ląstelėse atsiranda nuo pastojimo momento; dažni anomalijos yra RP1 genų (esant retinitui pigmentosa-1, autosominė dominuojanti) , RHO (RP4, autosominis dominuojantis) ir RDS (RP7, autosominis dominuojantis). Ne paveldimos pigmento retinito priežastys yra retos, tačiau galimybė rasti pavienį atvejį (savaiminė mutacija), kai nėra šeimos istorijos liga.
.jpg)